Eine neue Wirkstoffklasse für viele Indikationen?
Neurokinin-Rezeptor-Antagonisten
Steuerung. Neurokinine sind im Zentralnervensystem in Gehirn und Rückenmark sowie im peripheren und enteralen Nervensystem zu finden. Dabei handelt es sich um kleine, kurzkettige Neuropeptide, die aufgrund ihres ubiquitären Vorkommens bei der neuronalen Steuerung vieler physiologischer und pathophysiologischer Prozesse eine Rolle spielen. Zu diesen Prozessen zählen Nozizeption, Migräne, Erbrechen, Depression, Entstehung gastrointestinaler Reflexe sowie diverse entzündliche Prozesse.
Autoren: Mag. pharm. Dr. Karin Nemec, Univ.-Prof. Mag. pharm. Dr. Manfred Schubert-Zsilavecz

Tab. 1: Strukturen der in ersten Phasen der klinischen Prüfung für die Indikation Asthma untersuchten Neurokinin-Antagonisten
Die Eigenschaft der Neurokinine, eine rasche Kontraktion der glatten Muskulatur herbeizuführen, führte zu ihrer früheren Bezeichnung »Tachykinine«.
 |
Mag. pharm.
Dr. Karin Nemec |
Neben der Substanz P – sie ist der bisher am besten erforschte Neurotransmitter aus der Gruppe der Neurokinine – sind noch Neurokinin-A und Neurokinin-B genauer beschrieben. (Abb.1)
Neurokinine binden mit unterschiedlicher Präferenz an die bekannten Rezeptor-Subtypen. So bevorzugt Substanz P den NK1-, Neurokinin A den NK2- und Neurokinin B den NK3-Rezeptor. Die Neurokinin-Rezeptoren gehören zur großen Klasse der G-Protein-gekoppelte-Rezeptoren (GPCR). Die Existenz weiterer Subtypen neben den bekannten Neurokinin-1- (Substanz P-), Neurokinin-2- und Neurokinin-3-Rezeptoren wird vermutet. Die Proteine der bisher bekannten Rezeptoren bestehen aus 350 bis 500 Aminosäuren mit sieben hydrophoben transmembranären Domänen, die durch intra- und extrazelluläre Schleifen verbunden sind. Sie sind damit den adrenergen und muskarinergen G-Protein-gekoppelte-Rezeptoren sehr ähnlich.1
Als gesichert gilt, dass die Stimulierung des Neurokinin-1-Rezeptors eine Kontraktion der glatten Muskulatur, Gefäßerweiterung, glanduläre Sekretion (besonders der Speicheldrüsen), Extravasation von Plasmaproteinen und Erbrechen hervorruft. Die Aktivierung des Neurokinin-2-Rezeptors führt hingegen zur Kontraktion der glatten Muskulatur der Harnblase, bestimmter Darmabschnitte und Blutgefäße sowie zur Stimulierung von afferenten Nerven. Über den Neurokinin-3-Rezeptor ist bisher nur sehr wenig bekannt. Er vermittelt die Kontraktion begrenzter Darmareale und einiger weniger Blutgefäße. Neurokinin-Rezeptor-Antagonisten
Aufgrund der zahlreichen potenziellen Anwendungsgebiete von Neurokinin-Antagonisten, wie Schmerz- und Migränetherapie, Depression, Prävention und Therapie von Übelkeit und Erbrechen sowie entzündlicher Erkrankungen (Asthma bronchiale, Morbus Crohn) stellt diese neue Wirkstoffklasse ein interessantes Gebiet in der Arzneistoffentwicklung dar.
Die Erforschung der Neurokininwirkungen sowie das Design neuer Neurokinin-Rezeptor-Modulatoren ist allerdings nicht ganz unproblematisch.
Erhebliche Schwierigkeiten ergeben sich dadurch, dass große Unterschiede in der Neurokinin-Rezeptorprotein-Sequenz bei den einzelnen Spezies existieren, was wiederum Unterschiede in der Wirkung der Antagonisten zur Folge hat. Einige im Tierversuch vielversprechende Substanzen erwiesen sich bei ersten klinischen Untersuchungen am Menschen als unwirksam, da die Wirksamkeit von Neurokinin-Rezeptor-Antagonisten im Tiermodell keine Rückschlüsse auf eine Effektivität beim Menschen zulässt und somit positive Ergebnisse aus Tierversuchen nicht übertragbar sind.2
Des weiteren waren die ersten verfügbaren Neurokinin-Antagonisten peptidischer Struktur und nicht in der Lage, die Blut-Hirn-Schranke zu passieren. Weitere Nachteile waren ihre kurze Halbwertszeit und der hohe Syntheseaufwand.
Seit es gelungen ist, nicht-peptidische NK-Antagonisten zu synthetisieren, können rezeptorspezifische Wirkungen und physiologische Funktionen der Neurokinine durch diese pharmakologische Modellsubstanzen aufgeklärt werden. Die meisten dieser Wirkstoffe sind NK1-Antagonisten, aber auch NK2- und NK3-Antagonisten werden bereits hinsichtlich ihres therapeutischen Potenzials untersucht.
Die einzige Indikation, in der Neurokinin-Rezeptor-Antagonisten bisher Fuß fassen konnten, ist die Prophylaxe und die Therapie der Emesis.
Neurokinin-Rezeptor-Antagonisten und der Respirationstrakt
Neurokinin A und Substanz P werden mit einer Reihe von pathologischen Veränderungen in Lunge und Bronchien wie entzündlichen Prozessen, Plasmaextravasation, Bronchokonstriktion und Husten in Verbindung gebracht.
Der Hustenreiz kommt durch Stimulierung bestimmter sensorischer Nerven in den Atemwegen zustande. Zu diesen sensorischen Nerven gehören C-Fasern, die Substanz P, Neurokinin A sowie das calcitonin-generelatedpeptide (CGRP) enthalten. Neben ihrer Funktion bei der Weiterleitung von Informationen ins Hustenzentrum des Hirnstammes rufen sie durch die Neuropeptidfreisetzung eine Entzündungsantwort hervor, die durch Kontraktion der glatten Muskulatur und Ödeme gekennzeichnet ist und zu einer Verstärkung des Hustenreflexes führt.
Erste Tierversuchs-Studien mit den NK2-Rezeptor-Antagonisten Saredutant und dem NK1-Rezeptor-Antagonisten CP 99,994 zeigten eine antitussive Wirksamkeit, die durch zentralen Angriff ohne Beteiligung des Opioidmechanismus zustande kommt. Peripher verringern sie die Neurokinin-vermittelte Atemwegshyperreaktivität, die durch Entzündungsreaktionen und Bronchokonstriktion gekennzeichnet ist.
Im Falle von Asthma bronchiale führen Neurokinine zu einer Reaktion, die als neurogene Entzündung bekannt und die durch Vasodilation, Schleimsekretion, Plasmaproteinextravasation, Aktivierung der Leukozytenadhäsion und Bronchokonstriktion charakterisiert ist. Es scheint, als wäre Neurokinin-A der potentere Bronchokonstriktor als Substanz P. Substanz P führt zu vermehrter Schleimproduktion und mikrovaskulärer Leakage.
Erste klinische Untersuchungen mit den beiden oben genannten Wirkstoffen3,4 sowie dem NK1- und NK2-Rezeptor-Antagonisten FK-2245,6,7 und dem NK1-Rezeptor-Antagonisten FK-8888 bei asthmatischen Patienten zeigten jedoch nur sehr bescheidene klinische Effekte (Tab. 1, S. 616, im Einstiegsbild).
Neurokinin-Rezeptor-Antagonisten und der Gastrointestinaltrakt
Neurokinine spielen eine Rolle in der neuronalen Steuerung der gastrointestinalen Funktionen. Gebildet werden sie quantitativ am bedeutendsten in intrinsischen enterischen Neuronen, in extrinsischen primären afferenten Fasern, endokrinen Epithelzellen, Immunzellen und Endothelzellen. Die höchsten Konzentrationen an Substanz P und Neurokinin A finden sich im Darm, geringere Mengen in Ösophagus und Magen. Durch den Angriff an den drei verschiedenen Rezeptortypen beeinflussen sie sowohl das intestinale Nervensystem als auch die Muskulatur.
Neurokinine beschleunigen die Motilität in allen Schichten und Regionen des Magen-Darmtrakts und scheinen in allen seinen Abschnitten die Sekretion zu fördern. Die Substanz P wirkt an den Gefäßen des Gastrointestinaltrakts als Vasodilatator und erhöht die vaskulare Permeabilität.
Man geht daher davon aus, dass Neurokinine eine Rolle bei hypersekretorischen, vaskulären oder immunologischen Störungen des Gastrointestinaltrakts, bei gestörter Darmmotilität und somit bei entzündlichen Darmerkrankungen, bei Stressreaktionen, abdominalen Schmerzen und bei Darminfektionen spielen.9,10 Somit könnten Neurokinin-Antagonisten als spasmolytische, antidiarrhöische, antientzündliche und antinozizeptive Arzneimittel bei einer Vielzahl von Erkrankungen des Gastrointestinaltrakts, wie Morbus Crohn, Colitis ulcerosa, Dysmotilität oder Reizdarmsyndrom, von Bedeutung sein.3
Nicht vergessen werden sollte die Tatsache, dass bei allen Erkrankungen des Gastrointestinaltrakts – ob entzündliche oder funktionelle Störungen – viele Faktoren eine Rolle spielen und die Symptome durch das Zusammenspiel verschiedenster Mediatoren und Transmitter zustande kommen. Eine Therapie, die ausschließlich auf die Beeinflussung einer Substanz ausgerichtet ist, wird voraussichtlich nur bedingt erfolgreich sein. Dies mag auch der Grund sein, warum bis dato keine Ergebnisse über den Einsatz von Neurokinin-Rezeptor-Antagonisten bei Erkrankungen des Gastrointestinaltrakts vorliegen.
Neurokinin-Rezeptor-Antagonisten für den Einsatz im Zentralnervensystem
 Schmerz und Migräne Schmerz und Migräne
Für die Substanz P und Neurokinin A werden nozizeptive Eigenschaften sowie die Beteiligung an Entzündungsreaktionen postuliert.11 Man geht davon aus, dass sie als direkte Antwort auf schmerzhafte Stimuli freigesetzt werden und die körperlichen Antworten auf Schmerz beeinflussen.
Erste klinische Studien mit den Wirkstoffen CP-99,994 und Lanepitant zur Therapie postoperativer sowie osteoarthritischer Schmerzen zeigten jedoch keine oder nur sehr geringe Wirksamkeit.12,13
Zu einer Neubewertung der Rolle von Substanz P beim Schmerzgeschehen und einem Paradigmenwechsel in der Entwicklung neuer Analgetika führte die Tatsache, dass im Tierversuch der analgetische Effekt schwacher Morphindosen durch die gleichzeitige Gabe geringer Dosen von Substanz P deutlich erhöht war. Die Analgesie war aber ausschließlich auf die Aktivierung von Opioidrezeptoren zurückzuführen, da der pharmakologische Effekt durch Naloxon antagonisiert werden konnte. Somit bewirkt die Substanz P im Dorsalhorn die Freisetzung von endogenen Opioiden und reguliert die analgetische Aktivität des postsynaptischen Opioidsystems. Es scheint die Koaktivierung des Neurokinin-1-Rezeptors Vorraussetzung für die Opioidantworten zu sein.14,15
Das chimäre Peptid ESP7, das die amino-terminalen Aminosäuren des endogenen Opioids Endomorphin-2 und die c-terminalen Aminosäuren der Substanz P besitzt (siehe Abb. 2), ruft bei Ratten durch Angriff an m-Opioid- und Neurokinin-1-Rezeptoren Analgesie ohne Entwicklung einer Opioidtoleranz hervor. Blockiert man vor der Gabe von ESP7 die Substanz-P-Rezeptoren, werden sowohl eine starke Abnahme der analgetischen Potenz als auch Toleranzentwicklung festgestellt.16
Ob diese Erkenntnisse über das Schmerzgeschehen bei Tieren auf den Menschen übertragbar sind und wie sie Eingang in die weitere Entwicklung von Neurokinin-Modulatoren finden, bleibt abzuwarten.
Eine Migräneattacke ist charakterisiert durch die Dilatation kranialer Gefäße, der Änderung der neuronalen Aktivität des Trigeminuskerngebiets im Hirnstamm sowie durch Entzündungsreaktionen in den perivaskulären Anteilen von Duraarterien mit Freisetzung vasoaktiver und inflammatorischer Neuropeptide (CGRP, calcitonin-gene related peptide).17 Der akute Schmerz wird jedoch von keinem dieser Mechanismen allein ausgelöst, daher kommt es durch reine Vasokonstriktoren wie Adrenalin zu keiner Analgesie. Nur Wirkstoffe, die sich in den Tiermodellen für die durale Entzündung als wirksam erwiesen, bewirken eine Aufhebung des Migräneschmerzes (wie 5-HT1B/D-Agonisten, Triptane).
Obwohl als gesichert gilt, dass die Substanz P Extravasation, Plättchenaggregation und endotheliale Adhäsion weißer Blutkörperchen begünstigt, wodurch es in der Folge zur Bildung lokaler Ödeme kommt, zeigten die zwei nichtpeptidischen NK1-Rezeptor-Antagonisten, Lanepitant und Dapitant, in ersten klinischen Untersuchungen keine relevanten Effekte auf den akuten Migräneschmerz (Abb. 3).18,19
Depression
Der im Gehirn am stärksten exprimierte Subtyp ist der Neurokinin-1-Rezeptor. Er findet sich vor allem in Hirnarealen, die das affektive Verhalten und die neurochemischen Antworten auf Stress steuern, die höchsten Konzentrationen davon im limbischen System (in der Amygdala sind 10% der Neurone Substanz P-Neurone), im Corpus striatum und Hirnstamm.20
Erste klinische Studien mit dem Neurokinin-1-Rezeptor-Antagonisten Aprepitant, der aufgrund seiner hohen Affinität, Selektivität, oraler Bioverfügbarkeit und Wirkdauer als potentes Antidepressivum zur weiteren Untersuchung geeignet schien, lieferten positive Ergebnisse.21
Verglichen wurden 20 mg Paroxetin mit 300 mg Aprepitant einmal täglich. Die beiden Substanzen zeigten eine vergleichbare antidepressive Wirksamkeit, Aprepitant verursachte aber weniger Schlaflosigkeit und sexuelle Dysfunktion. Andere Nebenwirkungen wie Kopfschmerzen, Übelkeit und Somnolenz waren mild und transient. Eine anxiolytische Wirkung von Aprepitant konnte ebenfalls nachgewiesen werden.22
Ob und wie weit Veränderungen im Substanz P-Gehalt oder an den Neurokininrezeptoren in der Pathogenese von psychischen Erkrankungen eine Rolle spielen, muss noch geklärt werden. Die Erkenntnisse aus diesen fehlenden Untersuchungen könnten die Grundlage für die weitere Entwicklung neuer Neurokininantagonisten zur Therapie von Depressionen bilden.
Neurokinin-1-Rezeptor-Antagonisten in der antiemetischen Therapie des Zytostatika-induzierten Erbrechens
Das Grundproblem der antiemetischen Therapie liegt in der Komplexizität der zentralen Steuerung von Nausea und Erbrechen. Die Funktionen und Bedeutung der beteiligten Neurotransmitter sind noch nicht restlos geklärt. In Abhängigkeit vom emesis-auslösenden Stimulus werden unterschiedliche Neurotransmitter freigesetzt, sodass rezeptorspezifische Arzneimittel meist nur gegen eine Art des Erbrechens wirksam sind. Eine spezifische antiemetische Therapie scheint nicht möglich, da die Funktion des Brechzentrums über multiple Rezeptorsysteme mit unterschiedlichen Transmittern und Rezeptoren gesteuert wird. Von besonderer Bedeutung ist dies beim Zytostatika-induzierten Erbrechen (Abb. 4).
Obwohl mit der Wirkstoffklasse der 5-HT3-Rezeptor-Antagonisten (»Setrone«) sehr effiziente Arzneistoffe zur Prävention und Therapie des akuten Erbrechens nach Zytostatikagabe (Abb. 5) zur Verfügung stehen, die mit Dopaminrezeptor-Antagonisten und/oder Corticoiden kombiniert werden können, ist das verzögerte Erbrechen noch nicht gut beherrschbar. Es kommen zwar Corticoide, 5-HT3- sowie Dopamin-D1,D2-Rezeptor-Antagonisten zum Einsatz, sie zeigen allerdings bei vielen Patienten nicht die erhoffte Wirksamkeit. Denn anders als beim akuten Erbrechen, das durch die Zellzerstörung im Gastrointestinaltrakt durch Zytostatika zustande kommt, wodurch Serotonin in hoher Konzentration aus den Enterochromaffinzellen freigesetzt wird und durch Stimulierung von 5-HT3-Rezeptoren Übelkeit und Erbrechen auslöst, ist das verzögerte Erbrechen nicht auf eine Stimulierung durch Serotonin, sondern vor allem durch Neurokinin-1 (Substanz P) zurückzuführen.
Nachdem sich die NK1-Rezeptor-Antagonisten Vofopitant23 und CP 12272124 in der Therapie des postoperativen Erbrechens als wirksam erwiesen hatten, wurden erste Studien mit Neurokinin-1-Rezeptor-Antagonisten im Rahmen der Zytostatikatherapie durchgeführt.
Dabei wurde die Wirksamkeit eines Neurokinin-1-Rezeptor-Antagonisten als antiemetische Monotherapie sowie die Ko-Medikation von Neurokinin-1-Rezeptor-Antagonist plus 5-HT3-Rezeptor-Antagonisten und Dexamethason in der Prävention und Therapie der cisplatin-induzierten Emesis untersucht. Von den eingesetzten Wirkstoffen (wie Ezlopitant25 oder Vofopitant26) war die potenteste Verbindung das oral applizierbare Aprepitant.27,28,29,30,31
Wie zu erwarten, war die Wirksamkeit der Neurokinin-1-Rezeptor-Antagonisten in der Monotherapie der Kombination aus 5-HT3-Rezeptor-Antagonist (Granisetron oder Ondansetron i.v.) mit Dexamethason (peroral), die am effizientesten das akute, vor allem durch Serotonin ausgelöste Erbrechen verhindert, weit unterlegen. Allerdings konnte gezeigt werden, dass die antiemetische Potenz dieser Kombination durch Zugabe von Aprepitant noch gesteigert werden kann.
Das wichtigste Ergebnis dieser Untersuchungen war jedoch, dass das verzögerte Erbrechen, dessen Mediator in erster Linie die Substanz P darstellt, am besten mit Aprepitant – beziehungsweise der Kombination von Aprepitant mit Dexamethason – unterdrückt werden konnte.
Aprepitant
Aprepitant (Emend®) wurde vor kurzem für die Kombinationstherapie mit anderen Antiemetika zur Prävention der akuten und verzögerten Nausea und Emesis bei der Therapie mit hoch emetogenen Zytostatika, einschließlich hochdosiertem Cisplatin, zugelassen (Abb. 6).
Aprepitant ist ein selektiver Neurokinin-1-Rezeptor-Antagonist mit keiner bis geringer Affinität zu 5-HT3-, Dopamin- und Corticoid-Rezeptoren. Aprepitant ist in der Lage, die Blut-Hirn-Schranke zu passieren und verstärkt durch Blockade der zentralen NK1-Rezeptoren die antiemetische Wirkung der 5-HT3-Rezeptor-Antagonisten sowie von Dexamethason.
Die erste Gabe (125mg) muss etwa eine Stunde vor Verabreichung der Zytostatikatherapie erfolgen, gemeinsam mit der Kombination von Glucocorticoid/5-HT3-Rezeptor-Antagonist. (Abb. 7). Die gleichzeitige Nahrungsaufnahme hat auf die Pharmakokinetik von Aprepitant keinen Einfluss.
Zwei weitere Dosen von 80mg erfolgen am Morgen von Tag zwei und drei, gemeinsam mit dem Glucocorticoid.32 Da Aprepitant das CYP3A4 inhibiert und Glucocorticoide Substrate diese Isoenzyms sind, sollte die übliche Corticoiddosis um etwa 50 Prozent bei oraler und 25 Prozent bei parenteraler Gabe reduziert werden.33,34
Bei älteren Patienten, Patienten mit Niereninsuffizienz oder leichter bis mittelschwerer Leberinsuffizienz muss keine Dosisanpassung erfolgen. Für den Einsatz von Aprepitant bei Kindern liegen noch keine Daten vor.
Die orale Bioverfügbarkeit von Aprepitant beträgt 60 bis 65 Prozent. Mehr als 95 Prozent werden an Plasmaproteine gebunden, das Verteilungsvolumen beträgt 70L. Aprepitant unterliegt einem extensiven Metabolismus. Die Metaboliten zeigen eine nur schwache Aktivität. Die Ausscheidung erfolgt renal und biliär. Die terminale Halbwertszeit beträgt 9 bis 13 Stunden.35
Als häufigste Nebenwirkungen treten Somnolenz und Müdigkeit sowie gastrointestinale Effekte auf.
Aprepitant sollte aufgrund seiner Beeinflussung des Cytochrom P450-Systems nur mit besonderer Berücksichtigung etwaiger Interaktionen mit anderen Wirkstoffen eingesetzt werden. Dies gilt im Besonderen für jene Zytostatika, die über CYP3A4 abgebaut werden, zum Beispiel Docetaxel, Paclitaxel, Etoposid, Irinotecan, Imatinib sowie die Vinca-Alkaloide.
Bei der gleichzeitigen Verabreichung von Aprepitant mit Benzodiazepinen, die über CYP3A4 metabolisiert werden, wie Midazolam, Alprazolam oder Triazolam, ist mit einer Erhöhung der Plasmakonzentration der Benzodiazepine zu rechnen.
Bei Patienten, die eine Antikoagulantientherapie mit Cumarinen erhalten, sollte die Gerinnungszeit aufgrund der signifikanten Abnahme der International Normalized Ratio (INR) überwacht werden.
Bei Patientinnen, die orale Kontrazeptiva einnehmen, kann der Empfängnisschutz vermindert sein.
Als Inhibitor von CYP3A4 ist die Kombination von Aprepitant mit Pimozid, Terfenadin, Astemizol oder Cisaprid kontraindiziert.
Fazit
Obwohl in Fachkreisen in den letzten Jahren viel über den möglichen therapeutischen Nutzen von Neurokinin-Rezeptor-Modulatoren diskutiert wurde, sind die bisherigen Erfolge bei der Entwicklung klinischer relevanter Wirkstoffe doch eher bescheiden. Die Gründe hierfür sind vielfältig, wie lückenhaftes Grundlagenwissen, die fehlende Aussagekraft von Tierversuchen oder die klinische Prüfung von Wirkstoffen mit unpassendem pharmakokinetischem Profil. Trotz dieser Rückschläge bleibt die Entwicklung von potenten Neurokinin-Rezeptorantagonisten ein lohnendes Ziel der pharmazeutischen Industrie. Basis hierfür ist jedoch die vertiefte Kenntnis der Rolle von Neurokininen im pathologischen Geschehen der beschriebenen Krankheitsbilder und darauf aufbauend die Entwicklung von optimierten Wirkstoffen mit selektiven pharmakodynamischen Eigenschaften und angepassten pharmakokinetischen Wirkprofilen.
Der erste Wirkstoff, der diese Bedingungen erfüllt, ist Aprepitant zur Prävention und Behandlung des Zytostatika-induzierten Erbrechens, dessen vor kurzem erfolgte Einführung wesentlich zur Verbesserung der Lebensqualität onkologischer Patienten beitragen wird.
Abb. 1: Aminosäuresequenz und Rezeptorpräferenz der Neurokinine: 
Abb. 2: Sequenz und zweidimensionale Struktur von ESP7:

Abb. 3: Dapitant und Lanepitant: 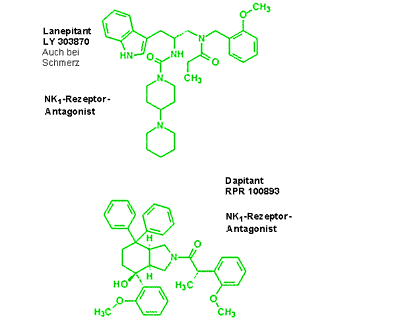
Abb. 4: neuronale Pfade und auslösende Faktoren für Zytostatika-induziertes Erbrechen 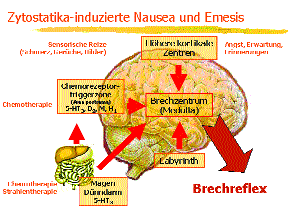
Abb. 5: Klassifizierung von Übelkeit und Erbrechen:

Abb. 6: Aprepitant: 
Abb. 7: Prophylaxe von Nausea und Emesis, basierend auf den Empfehlungen der American Society of Clinical Oncology: 
Literatur in der Redaktion Anschrift der Verfasser: Mag. pharm. Dr. Karin Nemec, Anstaltsapotheke Donauspital/SMZ-Ost, Langobardenstraße 122, !-1220 Wien, Tel.
+ 43 1 288 02 5127, E-Mail: karin.nemec@smz.magwien.gv.at
Univ.-Prof. Mag. pharm. Dr. Manfred Schubert-Zsilavecz, Institut für Pharmazeutische Chemie, Johann Wolfgang Goethe-Universität. Marie Curie-Straße 9, D-60439 Frankfurt am Main, Tel. +49 69 798 29339, E-Mail: Schubert-Zsilavecz@pharmchem.uni-frankfurt.de |